Introduktion
På 1950-talet kunde forskare visa att DNA är ärftlighetens molekyl. Det stod då klart att DNA består av en kod som ger alla celler instruktioner om bland annat vilka proteiner som ska tillverkas. Cellen behöver proteiner för att fungera.
Variation i DNA-sekvensen och därmed i uppbyggnaden av proteiner leder till olika egenskaper. En del egenskaper rör grundläggande funktioner inne i cellen medan andra är synliga för ögat, till exempel en blommas färg eller en människas längd.
Eftersom allt DNA kopieras till alla nya celler så går den genetiska koden i arv. Upptäckten av DNA som ärftlighetens molekyl var ett enormt steg för vetenskapen, men det skulle ta ytterligare några decennier innan den kunde avkodas.
År 1977 utvecklades parallellt två olika metoder för att sekvensera DNA. Med sekvensering kan forskare bland annat studera enskilda gener och jämföra DNA mellan arter. Det blev med sekvensering också möjligt att koppla samman genvarianter med olika egenskaper och med sjukdomar.
Både nya och äldre metoder används
Den metod som kom att användas mest av de två kallas för Sanger-sekvensering efter upphovsmannen Frederick Sanger, och används fortfarande rutinmässigt i många laboratorier. Metoden lämpar sig väl för att studera enstaka gener och mutationer.
Under de senaste två årtiondena har den tekniska utvecklingen inom DNA-sekvensering gått väldigt snabbt. Idag kan en organisms hela genom sekvenseras på bara några timmar med nya metoder som har samlingsnamnet Next Generation Sequencing (NGS).
Så här fungerar Sanger-sekvensering
Det DNA som ska sekvensbestämmas används som en mall för att tillverka nytt DNA som går att ”läsa”. Anledningen till att det går att läsa är att den nya DNA-sekvensen delvis är uppbyggd av nukleotider (med kvävebaserna A, T, C, G) som är märkta med en flurofor. En sorts fluorofor för varje nukleotid.
När en fluorofor träffas av ljus av en viss våglängd avger den i sin tur ljus (emitterar). Eftersom A, T, C, G är kopplade till olika fluoroforer, som avger ljus vid olika våglängd, så går det att läsa i vilken ordning de kommer i DNA-sekvensen. På datorskärmen visas det som ett kromatogram i olika färger som på bilden nedan.
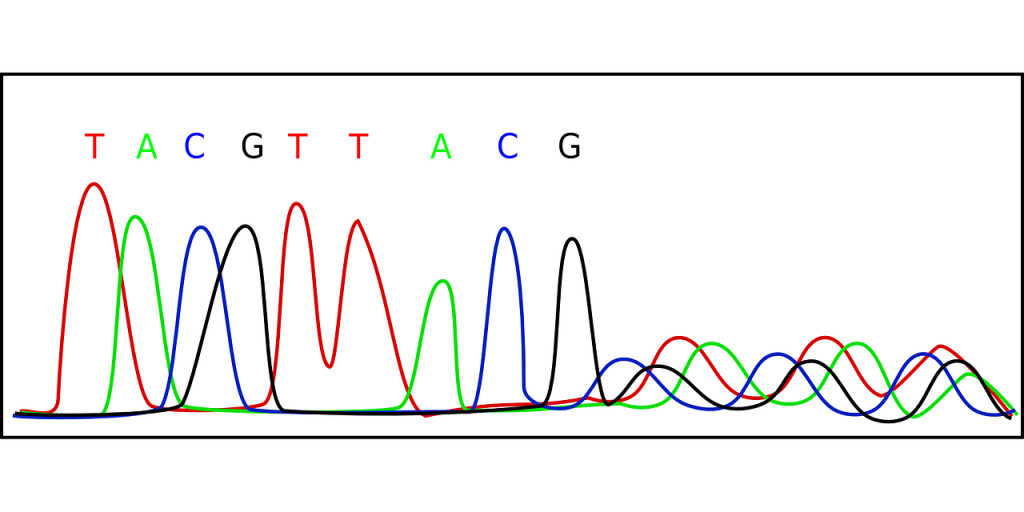
Hela sekvenseringsprocessen kan delas upp i tre steg:
Steg 1, reaktionerna bereds
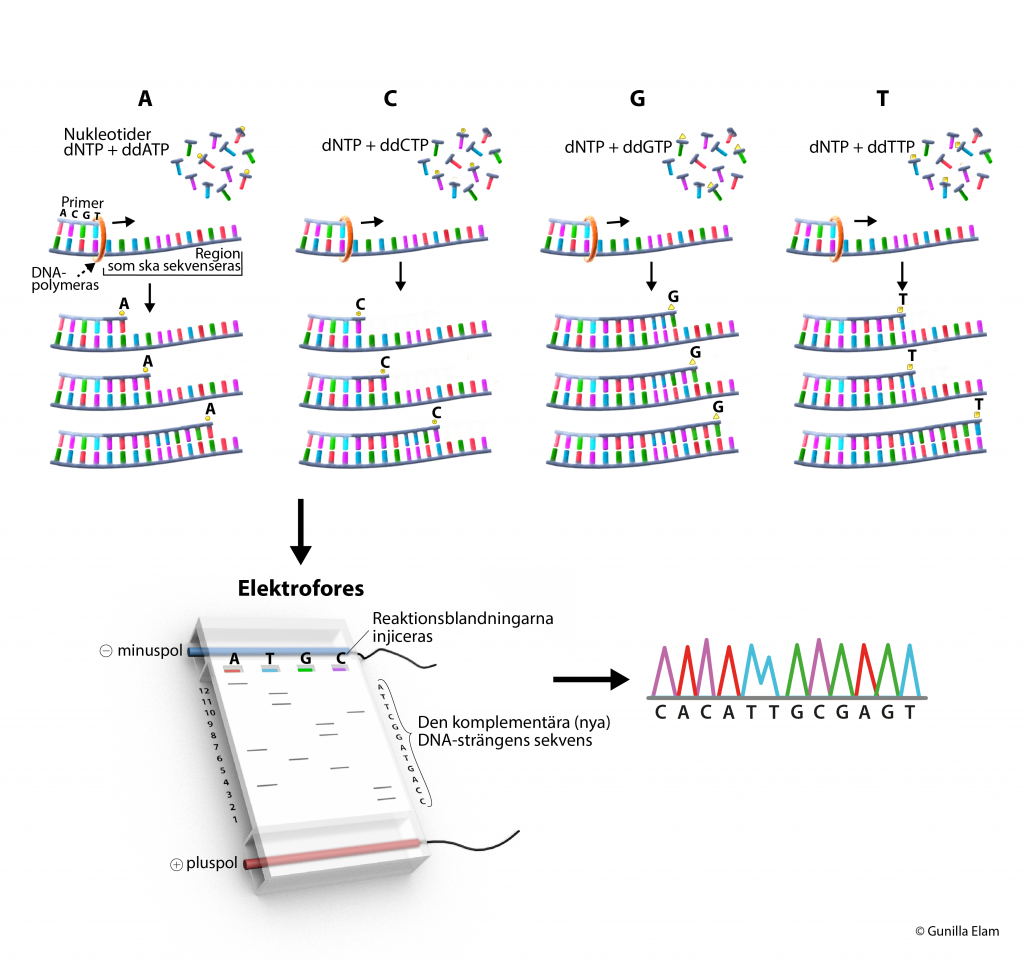
Fyra olika reaktionsblandningar bereds som innehåller:
- DNA (templat)
- Primers som visar vilken del av DNA-sekvensen som ska bestämmas.
- Enzymet DNA-polymeras som tillverkar den nya DNA-sekvensen.
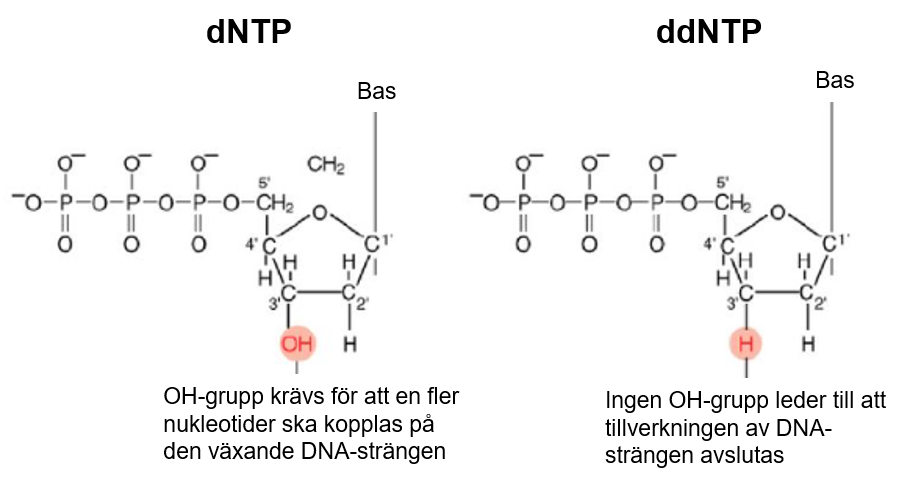
- Byggstenar i form av två olika slags fria nukleotider (dNTPs och ddNTPs) som under reaktionen frigörs till byggstenarna A, T, G, C. dNTPs (deoxynukleotidtrifosfater) är en mix av dATP, dTTP, dGTP, dCTP och tillsätts i alla reaktionsblandningar. ddNTPs (di-deoxynukleotidtrifosfater) är kopplade till en flurofor och i en av de fyra varje reaktionsrören tillsätts antingen ddATP, ddTTP, ddGTP eller ddCTP.
Steg 2, reaktionerna startar och nya DNA-sekvenser tillverkas
Tillverkningen av nytt DNA startar när de fyra reaktionsblandningarna placeras i en termocykler och ett PCR-program initieras med upprepade cykler av uppvärmning och nedkylning.
Då händer det här:
- Denaturering – Vid 94 grader C separeras de två strängarna i DNA-spiralen och DNA-sekvensen görs tillgänglig för primers och DNA-polymeras.
- Hybridisering – Mellan 55 – 68 grader C binder primers komplementärt till DNA och markerar startposition för DNA-polymeraset.
- Elongering – Vid 72 grader C börjar DNA-polymeras arbeta och bygger vidare på primer-sekvensen. En ny sträng DNA tillverkas när nukleotider som är komplementära till den motsatta strängen kopplas på en efter en.
De inmärkta nukleotiderna (ddNTPs) spelar en viktig roll!
I reaktionen är antalet nukleotider med fluorofor (ddNTPs) mycket färre i antal än de nukleotiderna utan (dNTPs). ddNTPs skiljer sig från dNTPs på två avgörande sätt.
- De har en OH-grupp mindre vilket gör det omöjligt för DNA-polymeras att koppla fler nukleotider efter en ddNTP. Det innebär att tillverkningen tar stopp när en ddNTP byggts in i den växande strängen och att alla nya strängar slutar med en byggsten som är kopplad till en fluorofor. De nya DNA-sekvenserna blir dessutom olika långa.
- De fyra olika nukleotiderna i ddNTP är kopplade till en fluorofor med olika ”färg” (återsänder ljus av olika våglängd) som gör att de går att särskilja.
Resultatet blir att i reaktionsröret där ddATP tillsattes finns en blandning av överlappande DNA-sekvenser av olika längd som alla avslutats med ett A. I reaktionsröret där ddTTP tillsattes finns en blandning av överlappande DNA-sekvenser som avslutas med T, och så vidare för de andra två reaktionsrören.
Steg 3, sekvenserna sorteras och analyseras
Innehållet i de fyra rören injiceras i en gelelektrofores som består av en gel av polymerer som bildar ett mikroskopiskt nät. När en strömkälla kopplas till gelen kommer DNA som är negativt laddad röra sig mot pluspolen som finns i slutet av gelen. Hur snabbt de olika DNA-sekvenserna rör sig genom gelen beror på deras storlek. På det här sättet separeras och sorteras alla DNA-sekvenser som genererats i steg 2.
I slutet av kapillären passerat DNA-sekvenserna laserstrålar och den fluorofor som är kopplad till sekvensens sista nukleotiden lyser upp. Ljussignalen registreras av en sensor och omtolkas av en mjukvara till ett kromatogram med fyra kurvor, en för varje nukleotid. Kromatogrammet visualiseras tvådimensionellt där x-axeln representerar positionen på den sekvenserade DNA-strängen och y-axeln motsvarande nukleotids intensitet.
Zooma in bilden nedan för att se hur det fungerar!
Så här fungerar Next generation sequencing (NGS)
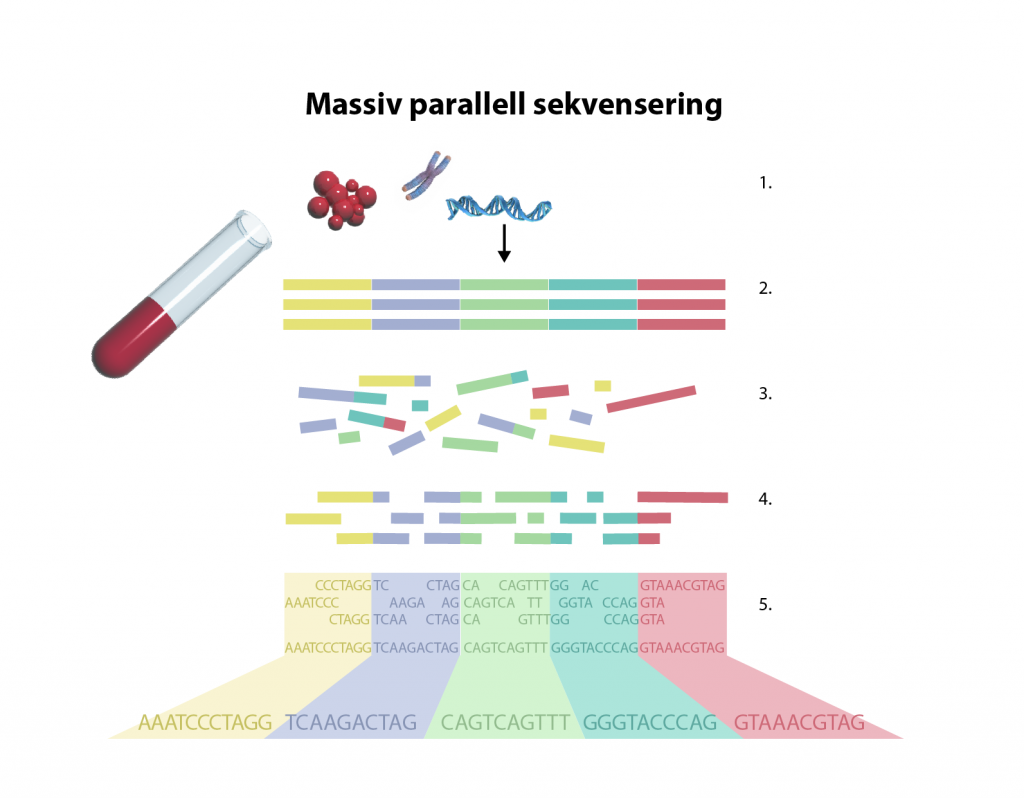
En NGS-metod som kallas massiv parallell sekvensering används till exempel för att sekvensera ett helt genom hos en organism. Det används också för att sekvensera alla exoner i alla gener, eller en panel av ett antal gener. Flera olika företag erbjuder så kallade plattformar för olika NGS. Det betyder att de tillverkar sekvenseringsmaskiner med tillhörande protokoll och mjukvara. De tekniska detaljerna skiljer sig åt mellan företagen men i stora drag går det till så här:
- DNA isoleras
- DNA fragmenteras, t.ex. med hjälp av starka ljudvågor (sonikering).
- DNA-fragmenten förbereds i ett steg som kallas biblioteksberedning (eng. library preparation). DNA-fragmenten kopplas samman (ligeras) med en sekvenserings-adapter som är specifik för maskinen som används och fästs på en fast yta (immobiliseras). Detta sker till exempel genom att DNA-fragmenten injiceras till en så kallad flödescell (se bild nedanför) där de fastnar på glasytans botten.
- DNA-fragmenten amplifieras så att de finns i många kopior.
- Alla DNA-fragment sekvenseras parallellt. Hur det går till skiljer sig mellan plattformar, till exempel tillverkas i en del system nytt DNA med DNA-polymeras och infärgade nukleotider (som vid Sanger-sekvensering). Ett annat sätt är att med enzymet DNA-ligas klistra in korta syntetiska infärgade DNA-fragment vars sekvens man känner till.
- De infärgade nukleotiderna, eller syntetiska DNA-fragmenten, läses av och omvandlas till information om varje enskild nukleotid.

De digitala sekvenserna som är cirka 100 – 200 baspar långa pusslas sedan ihop till en enda lång sekvens med hjälp av smarta algoritmer. Varje kvävebas har sekvenserats många gånger och man talar om sekvenseringens ”djup”, alltså hur många gånger i snitt en kvävebas lästs av.
Referensgenom är en viktig resurs
En viktig resurs vid sekvensering är att det finns ett referensgenom. Det är en digital sekvens som representerar alla kvävebaser i en arts genom i rätt ordningsföljd och där bland annat artens gener finns markerade. När en forskare sekvenserar DNA från till exempel en människa så finns den digitala resursen där som jämförelse. Forskaren kan då få reda på om det DNA som sekvenseras är en gen och i så fall vilken, och om sekvensen innehåller några genetiska varianter som inte finns i referensgenomet.
I andra databaser finns det information om hur vanliga olika genetiska varianter är och om den till exempel leder till en ändring i aminosyrasekvensen och proteintillverkningen.
Mycket kraft har lagts ner för att sekvensera så många arters genom som möjligt och tillgängliggöra den digitala sekvensen. Idag är nästan 100 000 arters genom sekvenserade och många finns tillgängliga digitalt i öppna databaser. Det är en oerhört viktig resurs för forskare, dels för att kunna studera enskilda arters genetik, men också för att jämföra olika arter. Till exempel är det via jämförelser av arters genom som forskare förstått att den del av genomet som inte innehåller gener, och som tidigare kallades för ”skräp-DNA”, har viktiga reglerande funktioner.
Exempel på vad man kan göra med sekvensdata:
- Diagnosticera genetiska sjukdomar molekylärt, det vill säga bekräfta att en patient har en mutation som man vet orsakar en viss sjukdom.
- Identifiera tidigare okända mutationer som orsakar en sjukdom.
- Identifiera källan till kontamination av mikroorganism, till exempel en bakterie, i en produkt.
- Karaktärisera gener som ger vissa bakterier resistens mot antibiotika.
- Diagnosticera infektionssjukdomar genom att sekvensera DNA (eller RNA) från ett angripande virus eller bakterie. Resultatet är vägledande för vilken behandling patienten ska få.
- Karaktärisera vilka bakterier som finns i tarmfloran hos en person.
- Konstruera fylogenetiska träd som visar hur olika arter är släkt med varandra.
Uppdaterad: 2024-06-24