Läkemedel är en produkt som är till för att förebygga, diagnosticera eller behandla sjukdomar hos människor och djur, och är samma sak som medicin eller farmaka.
Läkemedelsutvecklingens olika faser
All läkemedelsutveckling börjar med en lång så kallad pre-klinisk fas. I den fasen utvecklas läkemedlet och detaljerad kunskap införskaffas om läkemedlet och dess aktiva substansen med laboratorieexperiment.
Hur läkemedlets verkar testas först på odlade celler, sedan i försöksdjur och eventuellt vävnader. Om bevis finns för att en läkemedels ger önskad effekt så kan det under kontrollerade former prövas i människa. Det det kallas för kliniska studier eller kliniska prövningar.
För att få utföra en klinisk prövning i Sverige så krävs ett godkännande från Läkemedelsverket. En ansökan om att få genomföra en klinisk prövning genomgår alltid en etisk och vetenskaplig granskning av de pre-kliniska resultaten.
Den kliniska fasen är uppdelad i fyra.
- Fas 1. Läkemedlet prövas i låg dos i en grupp med friska frivilliga. Här undersöks läkemedlets säkerhet och hur det bryts ner i kroppen. Om läkemedlet bedöms tillräckligt säkert påbörjas nästa fas.
- Fas 2. Läkemedlet prövas i den grupp patienter som läkemedlet är utvecklat för. Här undersöks förutom säkerhet också behandlingseffekt och vilken dosering som är lämplig. Om läkemedlet bedöms säkert och att det verkar ge en positiv behandlingseffekt påbörjas nästa fas.
- Fas 3. Läkemedlet prövas i en större grupp patienter för att undersöka hur användbart det kommer vara. Läkemedlet jämförs med placebo eller andra behandlingsalternativ. Efter den här fasen kan utvecklaren ansöka om marknadsgodkännande.
- Fas 4. Läkemedlet har kommit ut på marknaden men data fortsätter att övervakas, till exempel samlas information om nya biverkningar in.
Specifik miljöriskanalys krävs för GMO-läkemedel
När ett GMO-baserat läkemedel ska prövas kliniskt inom EU ska GMO-lagstiftningen följas. Det innebär att en ansökan om att få genomföra en klinisk prövning även ska innehålla en specifik miljöriskanalys, en ERA (environmental risk assessment), i enlighet med EU-direktivet för avsiktlig utsättning av GMO i miljön (EU direktiv 2001/18/EG).
I direktivet för avsiktlig utsättning definieras en organism och en GMO så här:
Organism: varje biologisk enhet, som kan föröka sig eller överföra genetiskt material.
GMO: en organism, i vilken det genetiska materialet har ändrats på ett sådant sätt som inte sker naturligt genom parning och/eller naturlig rekombination.
Ansökningstiden är något längre för GMO-baserade läkemedel, 90 dagar jämfört med 60 dagar för ett andra läkemedel. Det beror på att andra svenska myndigheter, bland andra Gentekniknämnden och Naturvårdsverket, ska hinna yttra sig angående ansökan.
I en ERA ska i huvuddrag följande inkluderas:
- identifiering av egenskaper som kan orsaka allvarliga effekter i miljön
- värdering av möjliga konsekvenser av varje skadlig effekt
- värdering av sannolikheten att en händelse med skadlig effekt inträffar
- riskhanteringsstrategier
- bestämning av den övergripande risken av GMO:n
Det går att läsa mer om ansökningsprocessen för att få utföra en klinisk prövning av GMO-baserade läkemedel på Tillstånd, godkännande och kontroll (lakemedelsverket.se).
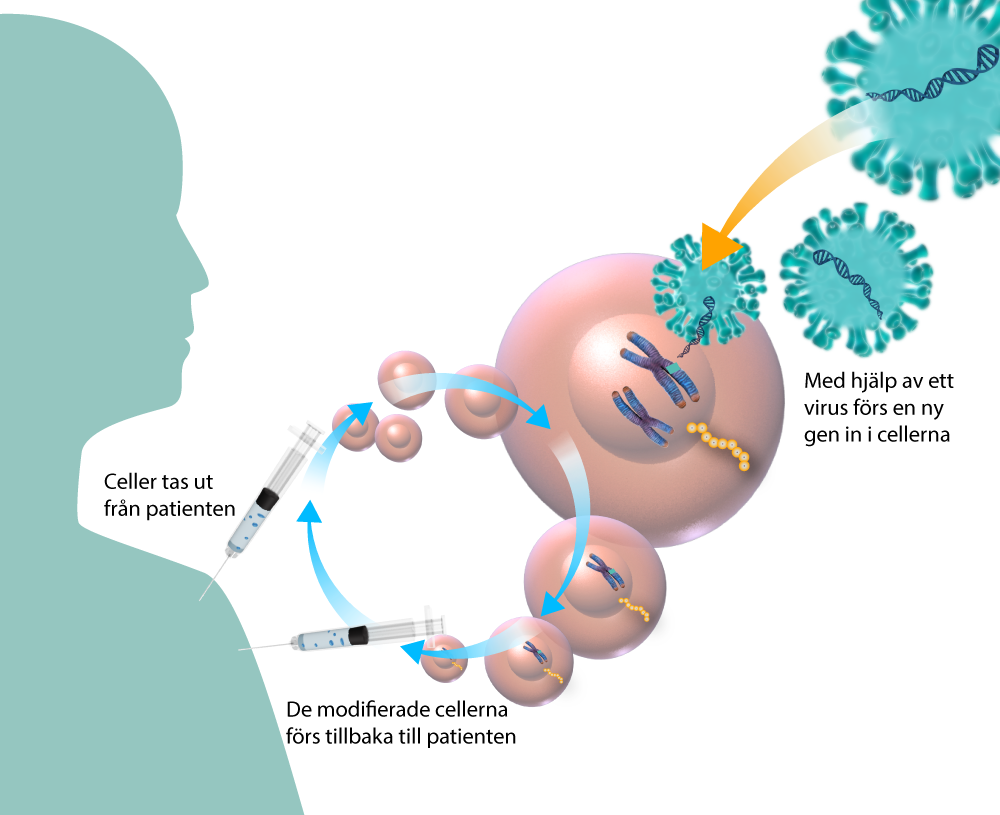
Exempel på GMO-baserade läkemedel
Det finns några olika kategorier av läkemedel som består av, eller innehåller GMO.
Genterapier med virusvektorer
Virus har en unik förmåga att ta sig in i sin värdorganisms celler och ända in i cellkärnan där majoriteten av allt DNA finns. Viruset fogar in sitt genetiska material (DNA eller RNA) i värdcellens DNA vilket leder till att värdcellen börjar tillverka virus-proteiner. Viruset själv har ingen egen proteintillverkning och kapar därför den funktionen hos värdcellen för att kunna spridas vidare.
Den här egenskapen hos virus har tagits tillvara inom forskningen. Till exempel används genetiskt modifierade virus vid genterapi för att transportera in en ny gen till en patients celler, istället för virusets DNA. Behandlingsstrategin är ofta att den nya genen ska kompensera för en gen som orsakar sjukdom. Viruset är modifierat så att det inte kan spridas och kallas för virusvektor, eller viral vektor.
Den virusvektor som använts oftast vid genterapi baseras på adeno-associerade virus (AAV). Den är utvald bland annat för att den i sin vildtypsform inte orsakar sjukdom hos människor. Även virusfamiljen lentivirus har visats vara en effektiv transportör i flera genterapier, i den familjen ingår till exempel HIV.

Genetiskt modifierade bakterier
Förutom genterapier finns några andra typer av läkemedel som regleras av GMO-lagstiftningen, till exempel bakterier som modifieras genetiskt och används som behandling. Ett sådant exempel är mjölksyrabakterier som modifierats så att de tillverkar ett mänskligt protein som stimulerar sårläkning.
Rekombinanta levande vaccin
En del vaccin som baseras på levande försvagade virus eller bakterier regleras också under GMO-lagstiftningen. Strategin bakom dessa vaccin är att viruset eller bakterien som vaccinet ska skydda mot ges i en så försvagad form att det inte orsakar sjukdom. Ibland odlas de länge i cellkultur vilket gör att de försvagas, och ibland används genteknik. I det senare fallet har viktiga gener för spridning tagits bort.
EU godkänner läkemedel som ska ut på den europeiska marknaden
Tillstånd för att få introducera ett nytt GMO-baserat läkemedel på marknaden i Sverige och inom övriga EU ges av EU-kommissionen efter rekommendation från EU:s läkemedelsmyndighet, EMA (European Medicine Agency). Det kallas för den centrala processen (CP) och är obligatoriskt för GMO-baserade läkemedel.
I ansökan om marknadsintroduktion ska bland annat resultaten från de kliniska prövningarna ingå som ska visa att läkemedlet är säkert och ger en positiv behandlingseffekt. I övrigt ställs samma krav som vid ansökan om att få utföra en klinisk prövning av GMO-läkemedlet.
Det görs två olika oberoende utredningar av en ansökan, i två olika EU-länder. Utredningarna kallas Rapp och CoRapp. Dessutom utför EMA och ytterligare ett EU-land en granskning. Processen har flera ”rundor” när utredare ställer svar till företaget som vill sälja läkemedlet och baserat på svaren följer ofta nya rundor med frågor. Alla medlemsländer har rätt att inkomma med kommentarer till utredningarna.
Introduktion i den svenska vården
Om ett läkemedel som godkänts av EU-kommissionen ska användas i Sverige bestäms efter en hälsoekonomisk utvärdering. I fallet med genterapier så är kostnaden för en behandling väldigt hög vilket bidrar till att det är en komplicerad utvärdering.
Mer information om hur utvärderingen går till finns på sidan om Godkända genterapier.
Uppdaterat 2024-07-26