Recessiv eller dominant nedärvning
För en monogen sjukdom är nedärvningsmönstret tydligt. Det innebär att man kan förutspå risken för att ett barn ärver en sjukdom från en förälder. Beroende på nedärvningsmönster kan monogena sjukdomar delas upp i recessiva och dominanta.
För att en recessiv sjukdom ska utvecklas måste mutationen finnas hos båda föräldrarna. De kromosomer i ett kromosompar som ett barn ärver från sina föräldrar ska alltså båda ha mutationen. Ärvs bara en mutation från en förälder är barnet en så kallad frisk bärare. Den sjukdomsalstrande mutationen riskerar då överföras till nästa generation om två friska bärare får barn.
För att en dominant sjukdom ska bryta ut räcker det med att mutationen finns på en av de två kromosomer i ett kromosompar som ärvs av förädlarna.
Autosomala och X-bundna sjukdomar
Monogena sjukdomar grupperas även in i autosomala eller X-bundna sjukdomar beroende på vilken sorts kromosom den sjukdomsalstrande mutationen finns. Människor har 46 kromosomer som bildar 23 kromosompar. Ett av de paren är könskromosomerna som heter X och Y. Flickor har två X-kromosomer och pojkar en av varje. Övriga fyrtiofyra kromosomer kallas för autosomala.
Anledningen till att det oftast talas om X- och inte Y-bundna sjukdomar är att Y-kromosomen har få gener och väldigt få kända genetiska sjukdomar är därför kopplade till Y. En genetiska sjukdom som härrör från en mutation på X-kromosomen har ett annorlunda nedärvningsmönster än en autosomal sjukdom.
Det är främst för de monogena sjukdomarna som det finns hopp om att genterapi ska kunna användas som behandlingsform. Vid genterapi förs en ”frisk” genkopia in i patientens celler med syfte att ersätta den genkopia som bär den sjukdomsalstrande mutationen. För vissa sjukdomar, till exempel muskeldystrofi, finns redan en godkänd genterapi.
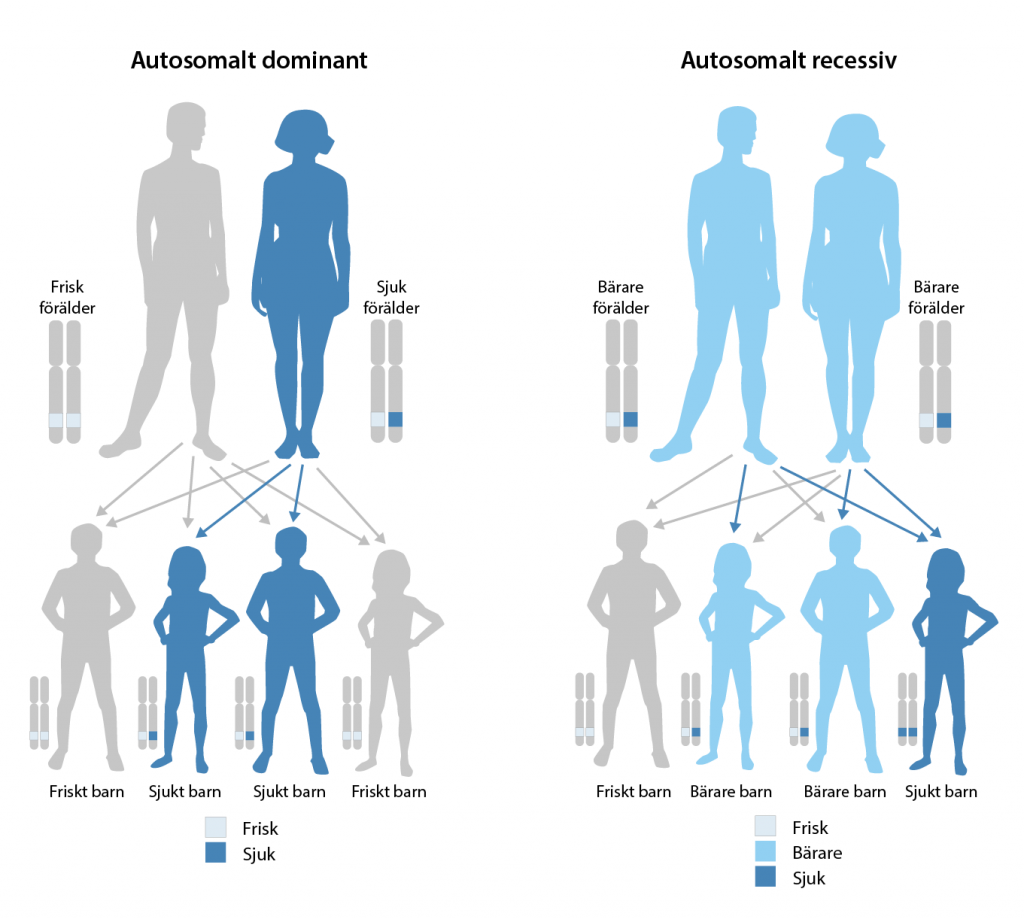
Illustration och copyright: Gunilla Elam.
Tre monogena sjukdomar:
Sicklecellanemi
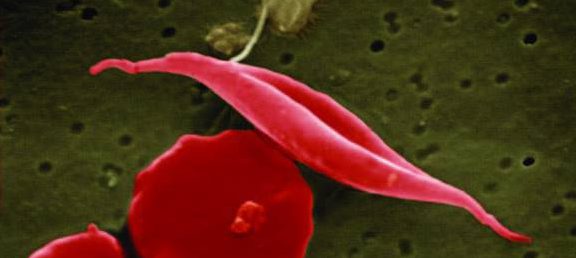
Sicklecellanemi är en autosomalt recessiv sjukdom som orsakas av en mutation i betaglobingenen, HBB, som finns på kromosom 11. Mutationen måste ha ärvts av båda förändrarna för att sjukdomen ska bryta ut helt. Betaglobin är en viktig beståndsdel av hemoglobin som sköter syretransporten i de röda blodkropparna.
Mutationen i betaglobin-genen leder till att hemoglobin blir felaktigt sammansatt och en del av de röda blodkropparna antar formen av en skära (eng. sickle). De deformerade röda blodkropparna klumpar ihop sig och kan bilda proppar i blodflödet och syrebrist i kroppens vävnader och organ. En patient med sickelcellanemi har symtom som till exempel blodbrist, akut smärta, bensår och njurskada.
Behandlingen består bland annat i smärtstillande läkemedel och i vissa fall stamcellstransplantation.
Det finns en genterapi för behandling av sicklecellanemi som är godkänd inom EU. Det är den första CRISPR/Cas9-baserade behandlingen som godkänts i världen.
Mer information om hur behandlingen fungerar finns i forskningsnyheten Världens första CRISPR/Cas-baserade genterapi godkänd inom EU.
Leukodystrofier
Det finns en grupp neurologiska sjukdomar som tillsammans kallas för autosomala dominanta leukodystrofier (ADLD). De orsakas av mutationer i en gen, men hos olika patienter kan det vara olika gener som ligger bakom. Sjukdomarna har ett dominant nedärvningsmönster vilket innebär att det bara krävs en mutation på en kromosom för att sjukdom ska utvecklas.
Gemensamt för alla ADLD är att den vita substansen, myelinet, som finns kring nervtrådarna i hjärnan och ryggmärgen skadas och förtvinar. ”Leuko” är det grekiska ordet för vit och ”dystrofi” betyder förtvining. När myelinet förtvinar leder det till nedsatt nervfunktion. Tidigt i sjukdomsförloppet påverkas icke-viljestyrda nervsystemet (t.ex. blodtryck, tarm- och urinblåsfunktion) och senare även de viljestyrda (använda olika muskler, kunna koordinera rörelser, tugga, svälja).
Det flesta fall av ADLD uppkommer i barndomen och har ett mycket allvarligt förlopp. Det finns även former som debuterar i vuxen ålder och av dessa är LMNB1-relaterad ADLD troligtvis den vanligaste i Sverige. Sjukdomen orsakas av en typ att mutation som kallas för duplikation som innebär att en DNA-sekvens, i det här fallet genen LMNB1, har fördubblats.
LMNB1 finns på kromosom 5 och kodar för proteinet lamin. Duplikationen gör att för mycket lamin tillverkas, och det leder i sin tur till att det tillverkas mindre mängd myelin. Resultatet blir att nervceller får en brist på myelin och en sämre förmåga att skicka nervimpulser.
Blödarsjukan (Hemofili A och B)
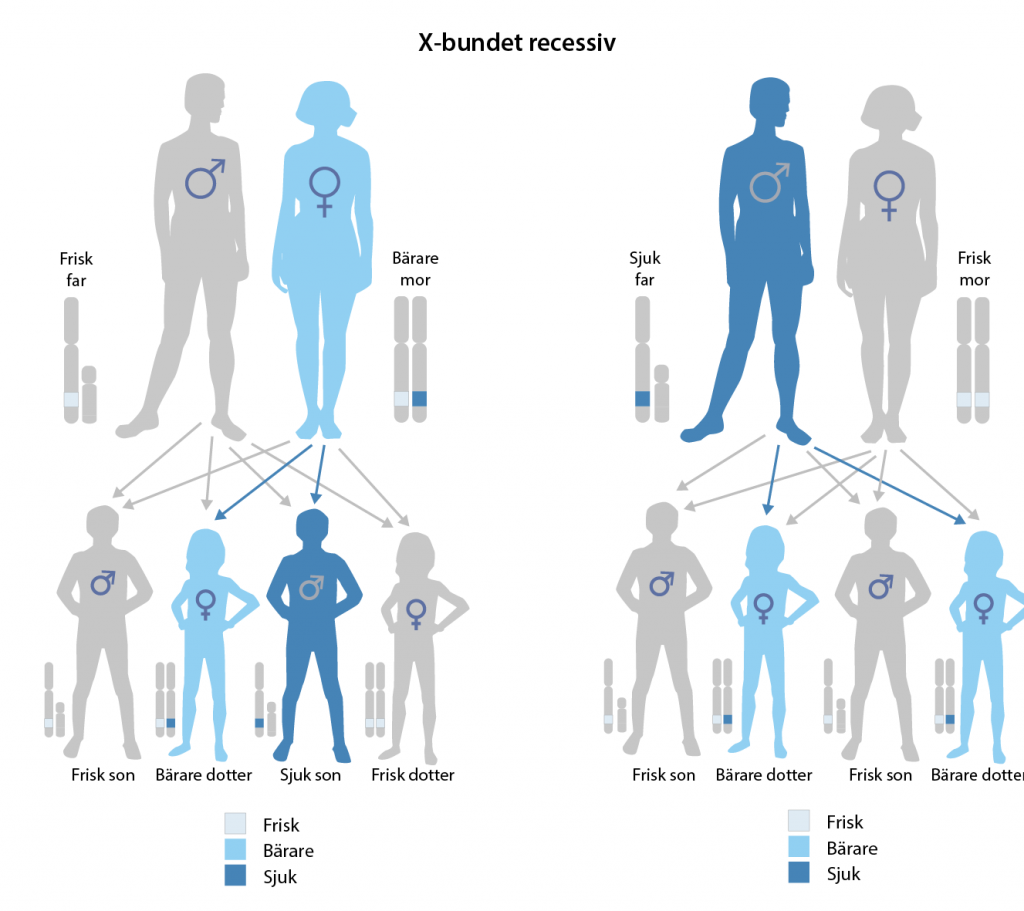
Hemofili A och B är två former av blödarsjuka som båda är X-bundna recessiva sjukdomar. De orsakas av mutationer i generna F8 (hemofili A) och F9 (hemofili B) som båda finns på X-kromosomen.
F8 och F9 kodar för koagulationsfaktorer som är viktiga för att blodet ska koagulera och bland annat hindra blodförlust vid skador. Hemofili förekommer i stort sett bara hos män eftersom de bara har en X-kromosom. Med sina två X-kromosomer har kvinnor oftast en ”frisk” genkopia som gör att koagulationsfaktorerna tillverkas som vanligt.
Hemofili kan vara en sjukdom med lindriga, medelsvåra eller svåra symtom. Hos en patient med medelsvår eller svår sjukdom uppstår blödningar lätt i leder och muskler. Blödningarna uppkommer till synes spontant, eller efter mindre skada, och är ofta mycket smärtsamma. Om blödningarna inte förebyggs orsakar de så småningom bestående förändringar i ledkapsel, ledbrosk och ben som med tiden kan ge funktionsnedsättningar och kronisk smärta.
I Sverige, och på många andra håll i världen behandlas barn som föds med blödarsjuka redan från tidig ålder. Behandlingen går ut på att injicera ett koncentrat av de koagulationsfaktorer som saknas. Koncentratet behöver ges flera gånger i veckan men har visat sig förhindra skador på leder och dessutom förbättra livskvaliteten för patienterna.
Det finns några genterapier som godkänts inom EU för behandling av hemofili A och B. Mer information finns på sidan om Genterapier.
Uppdaterad: 2024-03-27