Idag beviljade EU-kommissionen ett villkorat godkännande för genterapin Casgevy. Det är den första CRISPR/Cas9-baserade behandling som når patienter utanför kliniska prövningar. Samma behandling har godkänts i Storbritanniens och i USA under de sista månaderna av 2023.
Livshotande ärftlig blodsjukdom
Genterapin är utvecklad för blodsjukdomarna β -thalassemi och sicklecellanemi som orsakas av mutationer i en av de gener som kodar för uppbyggnaden av hemoglobin. I de röda blodkropparna ansvarar hemoglobin bland annat för att transportera syre till kroppens alla vävnader.
Den mutation som ger sicklecellanemi gör att vissa röda blodkropparna är defekta och deras form påminner om en månskära. Det är så sjukdomen har fått sitt namn – skära översätts med sickle på engelska. De defekta blodkropparna gör blodet mer trögflytande och proppar bildas som riskerar leda till akut blod- och syrebrist i olika delar av kroppen. Personer med sicklecellanemi upplever mycket smärta och lever i ständig risk för organsvikt.
Den mutation som personer med β -thalassemi har gör att en del röda blodkroppar skadas och går sönder vilket ger blodbrist. Hos de personer som är allvarligast sjuka försöker kroppen kompensera med att bilda mer blod som hjärtat måste pumpa runt i kroppen, utan att det förbättrar syresättningen. Det kan leda till hjärtsvikt.
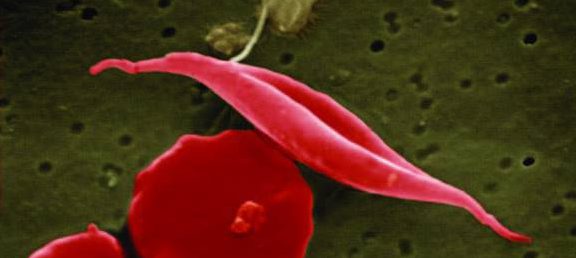
Patientens stamceller programmeras om med CRISPR/Cas9
Vid genterapin isoleras patientens egna blodbildande stamceller och redigeras med CRISPR/Cas9, innan de återförs som behandling. Själva redigeringen går ut på att stänga av en gen som heter BCL11A som fungerar som en broms för tillverkningen av så kallat fetalt hemoglobin. När CRISPR/Cas9 stänger av BCL11A så släpper bromsen.
Under fostertiden är det fetalt hemoglobin som sköter syretransporten i blodet men ungefär tre månader efter födseln slår tillverkningen om till ”vuxet” hemoglobin. Skillnaden mellan de olika formerna är inte så stor och fetalt hemoglobin har visats sig kunna kompensera för ett icke-fungerande vuxet hemoglobin.
Smärtfria efter behandling
I de kliniska prövningar som ligger till grund för godkännandet ingick ett hundratal patienter. Hos samtliga kunde normala mängder fungerande hemoglobin uppmätas i blodet efter behandlingen. Majoriteten av hemoglobinet var av den fetala formen.
Ingen av de behandlade patienterna med sicklecellanemi upplever längre de svåra episoder av smärta de tidigare behövde behandlas för och de mår generellt mycket bättre. Patienterna med β -thalassemi är inte längre i behov av blodtransfusioner. Flera patienter som uttalat sig i media säger att de fått ett nytt liv.
Det återstår att se om behandlingen kommer att användas inom några EU-länder. På prislappen som företagen satt på en dos står det 2,2 miljoner USD (cirka 23 miljoner SEK).
/Mia Olsson (2024-02-13)
Källor:
Pressmeddelande på företaget Vertex webbsida: European Commission Approves First CRISPR/Cas9 Gene-Edited Therapy, CASGEVY™ (exagamglogene autotemcel), for the Treatment of Sickle Cell Disease and Transfusion-Dependent Beta Thalassemia (vrtx.com)
Frangoul et al., CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. NEJM Dec 5 2020